AIDDISON
AIDDISON™ is Merck KGaA's first AI solution (SaaS platform) integrating drug discovery and synthesis. Combining generative AI (REINVENT), machine learning, and computer-aided drug design (CADD) tools, it enables medicinal chemists to rapidly perform virtual screening, scaffold hopping, lead compound identification, and optimization across a space of over 60 billion compounds. The platform is trained on over 20 years of experimentally validated pharmaceutical R&D data, and evaluates molecular synthesizability in real time via the SYNTHIA™ retrosynthesis API — the only platform in the industry that simultaneously addresses "what molecules to design" and "how to synthesize them."
Platform Overview
AIDDISON platform architecture overview — integrating generative AI, ADMET prediction, chemical space search, molecular docking, and synthesizability evaluation via SYNTHIA retrosynthesis API
Workflows implemented in AIDDISON — including molecular generation, property prediction, virtual screening, and retrosynthesis evaluation steps
Core Modules
| Module | Description |
|---|---|
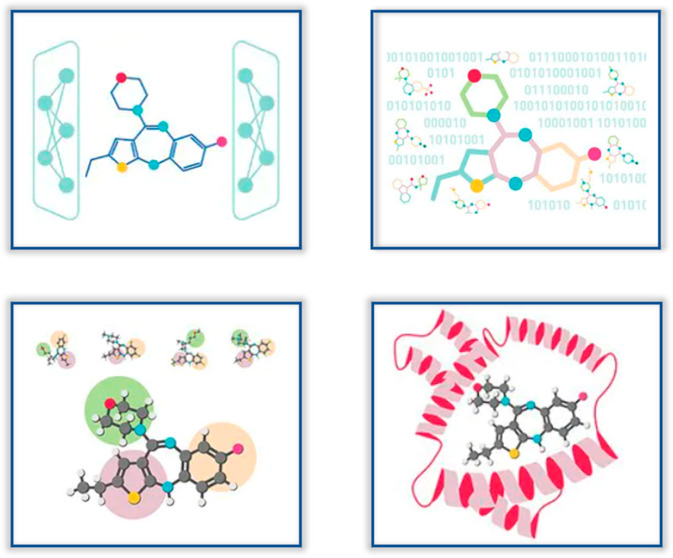
| De Novo Molecular Design (REINVENT) | Design novel molecules based on user-defined structural similarity and synthesizability, optimizing QED, ADMET properties, and binding affinity |
| ADMET Prediction | Predict drug absorption, distribution, metabolism, excretion, and toxicity properties |
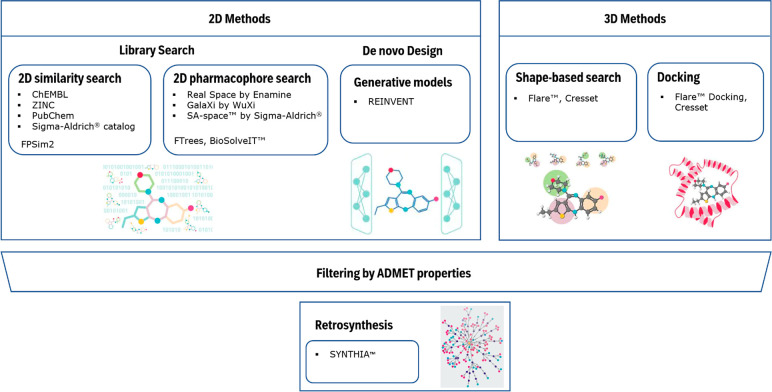
| Similarity & Druglikeness Search | Search over 60 billion compounds using FPSim2 / BioSolveIT FTrees algorithms, including SA-Space 25 billion+ virtual compounds |
| Molecular Docking | Visualize interactions between target molecules and proteins using Cresset Flare, and evaluate binding affinity |
| 3D Shape Matching | Align 3D molecular structures to reference ligands using Cresset Flare for scaffold hopping design |
| SYNTHIA Retrosynthesis | Evaluate molecular synthesizability and plan synthesis routes |
Module Illustrations
De Novo Molecular Design — REINVENT powered generative AI molecular generation
Similarity & Druglikeness Search — rapid screening across 60 billion+ compound space
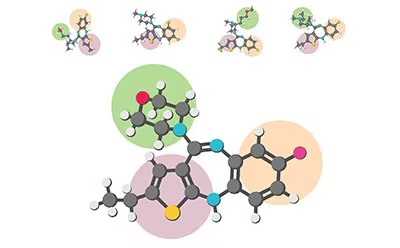
3D Shape Matching — Cresset Flare molecular structure alignment and scaffold hopping
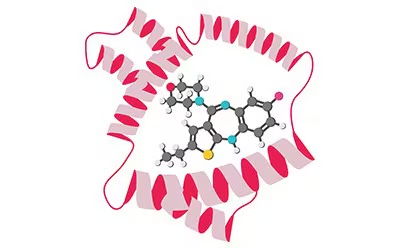
Molecular Docking — protein-ligand complex visualization and binding evaluation
Key Features
Integrated AI/ML and CADD Tools
Combines generative models, ADMET property prediction, large-scale chemical space search, and molecular docking in a single platform, eliminating the need to switch between multiple tools
60 Billion+ Compound Database
Screen from over 60 billion compound possibilities, identifying candidate molecules with key drug properties such as non-toxicity, solubility, and in vivo stability
SYNTHIA Retrosynthesis Integration
Optimal molecular designs can be sent directly to SYNTHIA retrosynthesis software to evaluate synthesizability and identify required reagents, bridging virtual design and actual manufacturing — an industry first
20+ Years of Experimentally Validated Data
Trained on over two decades of experimentally validated pharmaceutical R&D datasets, ensuring prediction reliability — not merely learning from literature or theoretical data
Secure and Compliant Web Platform
Compliant with ISO 27001 standards, providing the highest level of digital product information security certification, with strict protection of user research data and intellectual property
Low Learning Curve
Clean interface, automated complex tasks, seamlessly integrated ML models — enabling medicinal chemists without AI expertise to easily leverage advanced AI/ML tools
CADD Tool Visualization
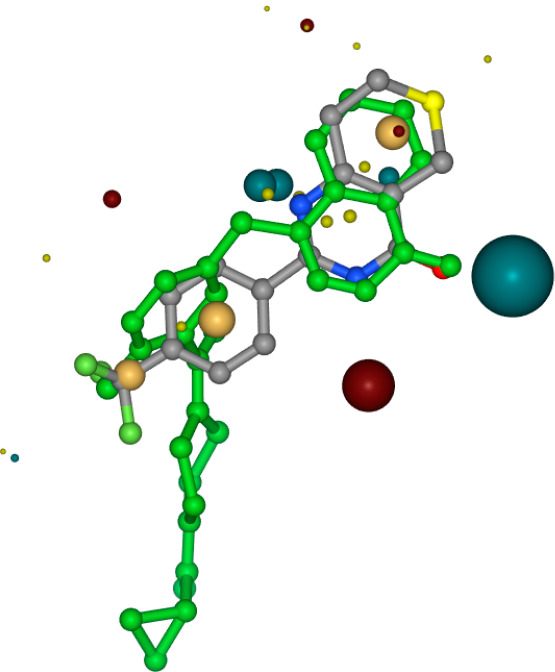
Shape-based alignment results for Olaparib — used for scaffold hopping, finding candidate molecules with similar shapes but novel structures from known active molecules
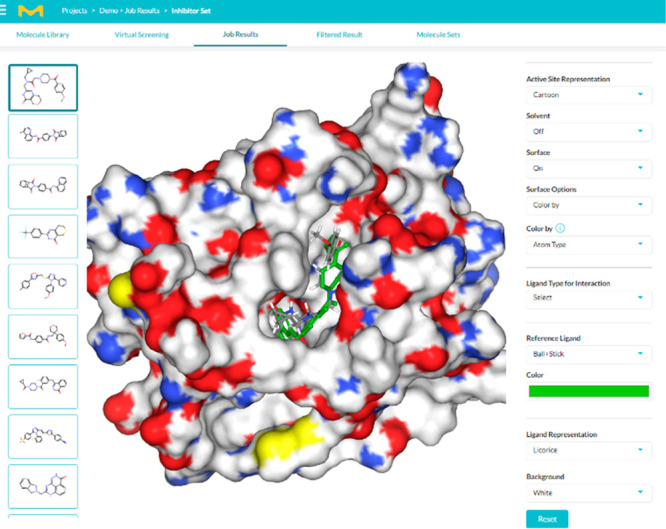
Molecular docking workflow result visualization — binding modes and docking scores of candidate molecules with target proteins
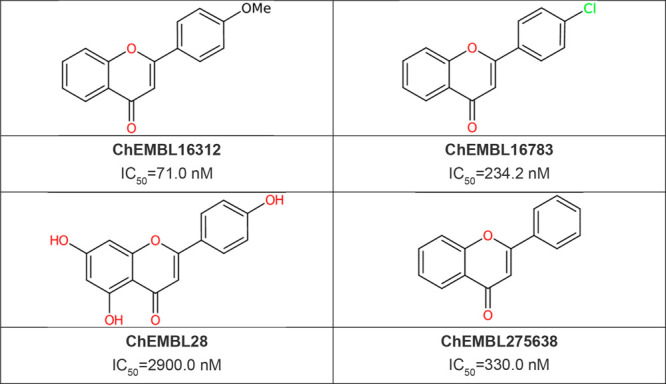
Case Study: Tankyrase Inhibitor
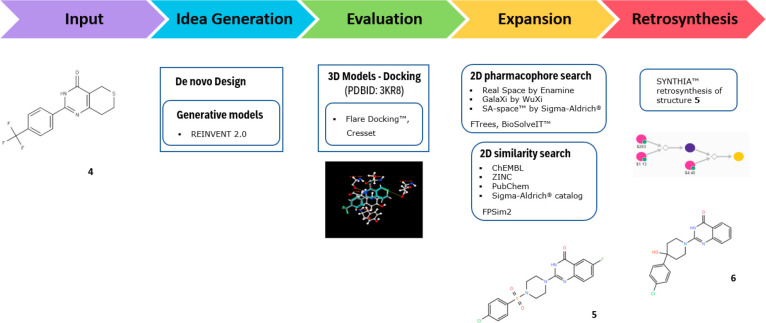
Tankyrase inhibitor design and optimization — starting from known flavonoid-type inhibitors, discovering new candidate molecules through AI generation, screening, and optimization
Chemical space overlap analysis of virtual compound libraries — comparing chemical space distributions of 3D shape search (green) and 2D pharmacophore search (gold)
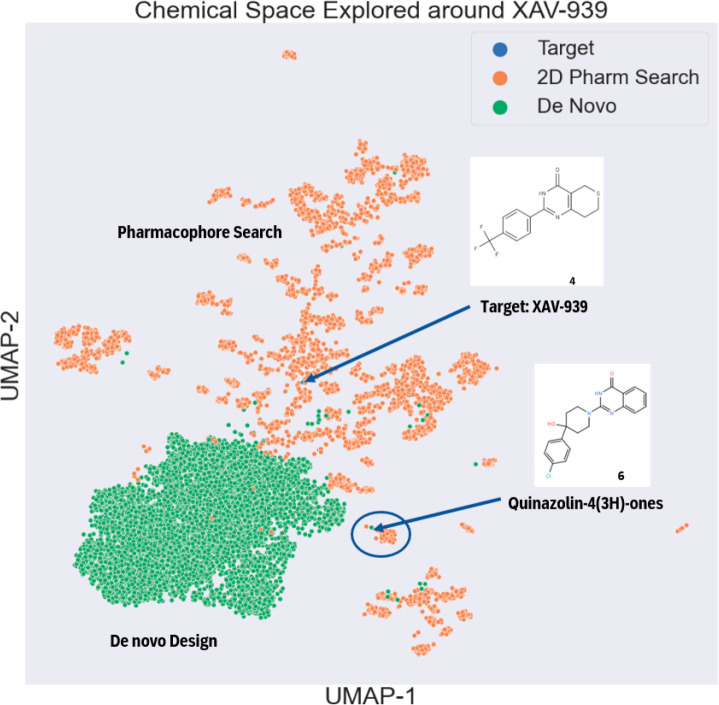
Pharmacophore search and chemical space exploration detailed results — demonstrating the platform's capabilities in large-scale compound screening
Position in the Drug Development Lifecycle
| Development Stage | AIDDISON Role |
|---|---|
| Post-Target Validation / Virtual Screening | Screen candidate molecules with desired properties from 60 billion+ compound space |
| Lead Compound Identification | Identify initial active compounds through molecular docking, shape matching, and pharmacophore search |
| Scaffold Hopping | Discover structurally novel molecules with similar pharmacological activity based on known active molecules, overcoming patent limitations |
| Lead Compound Optimization | Optimize candidate molecule drug properties using ADMET prediction and generative models |
| Synthesis Feasibility Assessment | Evaluate molecular synthesizability in real time during the design stage through SYNTHIA integration |
Technical Specifications
| Item | Specification |
|---|---|
| Platform Type | SaaS (Software as a Service) |
| Deployment | Cloud-based Web Platform |
| Security Certification | ISO 27001 |
| Compound Search Space | 60 billion+ compounds |
| Training Data | 20+ years of experimentally validated pharmaceutical R&D datasets |
| Retrosynthesis Integration | SYNTHIA Retrosynthesis API |
| Release Date | December 2023 |
| Developer | Merck KGaA, Darmstadt, Germany |
Application Areas
Pharmaceutical Industry
Accelerate virtual screening, lead compound identification, and optimization in drug discovery.
Biotechnology
Provide enterprise-grade AI drug discovery capabilities for small and medium biotech companies without building an in-house AI team.
Contract Research Organizations (CRO)
Rapidly perform virtual screening and candidate molecule evaluation for clients.
Academic Institutions
Medicinal chemistry and computational chemistry research, lowering the learning barrier for AI/ML technology.
Key Differentiators
Unified Design + Synthesis
The only platform in the industry that simultaneously addresses "what molecules to design" and "how to synthesize them," enabled through SYNTHIA integration
60 Billion+ Compound Space
Search scope far exceeds traditional compound libraries (typically millions to billions), vastly expanding drug discovery possibilities
20 Years of Experimentally Validated Data
Trained on experimentally validated pharmaceutical R&D data, not merely learning from literature or theoretical data, ensuring prediction quality
Low Learning Curve + ISO 27001 Security
Enables medicinal chemists without AI expertise to use advanced AI/ML tools, while protecting user intellectual property
Related Products
Design Next-Generation Drug Molecules with AI
Contact us to learn how AIDDISON can accelerate your drug discovery workflow, from virtual design to actual synthesis in one platform
Contact Us to Learn More