BIOVIA Materials Studio
Materials Science Modeling & Simulation Platform — A multiscale materials simulation environment from quantum to mesoscale

BIOVIA Materials Studio is a complete modeling and simulation environment that enables researchers in materials science and chemistry to predict the relationships between a material's atomic and molecular structure and its properties and behavior, in order to develop new materials. It is the world's most advanced and easy-to-use materials modeling platform, covering quantum, atomistic, mesoscale, statistical, and analytical simulation methods.
With Materials Studio, researchers across industries can design better materials of all types, including pharmaceuticals, catalysts, polymers and composites, metals and alloys, batteries and fuel cells, nanomaterials, semiconductors, and more. The platform supports an "in silico first" approach — reducing cost and time associated with physical testing and experimentation by virtually screening candidate material variants.
Version 1.0 released in June 2000, celebrating 25th anniversary in 2025
All of the world's top 25 pharmaceutical, biotech, and chemical companies are customers
Peer-reviewed publications citing Materials Studio (as of 2025)
Module Overview
Quantum Mechanics Tools
DFT-based solvers and semi-empirical methods for predicting properties from electronic structure.
| Module | Description |
|---|---|
| CASTEP | Plane-wave DFT code for simulating solids, interfaces, and surfaces — band structure, density of states, and optical properties of ceramics, semiconductors, and metals |
| DMol3 | DFT code for simulating electronic structure and properties of organic/inorganic molecules, molecular crystals, covalent/metallic solids, and infinite surfaces |
| ONETEP | Linear-scaling DFT code for accurate first-principles calculations on systems of up to thousands of atoms |
| DFTB+ | Semi-empirical module based on DFT tight-binding method, achieving quantum mechanical accuracy on larger systems |
| VAMP | Semi-empirical molecular orbital methods for rapid prediction of physical and chemical properties of molecular systems |
| QMERA | QM/MM method combining quantum calculation accuracy with force field calculation speed |
| FlexTS | Robust transition state search tool for identifying minimum energy paths between reactants and products |
| NMR CASTEP | First-principles prediction of NMR chemical shifts and electric field gradient tensors |
| KINETIX | Simulates chemical and physical adsorption, desorption, and diffusion on surfaces — catalyst activity and surface coverage |
| CANTERA | Chemical rate equation solver with reaction rates computable by DMol3 |
Classical Simulation Tools
Molecular dynamics, lattice dynamics, Monte Carlo methods, and force fields.
| Module | Description |
|---|---|
| Forcite Plus | Molecular mechanics and dynamics for predicting mechanical properties, diffusion coefficients, density changes. Supports GPU acceleration and COMPASS, CVFF, PCFF force fields |
| GULP | Materials optimization, property calculations, and dynamics; includes force fields for metals, oxides, semiconductors, and force field fitting tools |
| Amorphous Cell | Builds representative models of complex amorphous systems and predicts key properties |
| Sorption | Predicts fundamental adsorption and separation properties — adsorption isotherms and Henry's constants |
| Adsorption Locator | Finds low-energy adsorption sites for molecules on periodic and non-periodic substrates |
| Blends | Predicts phase diagrams, interaction parameters, and phase equilibria of mixtures |
| Conformers | Conformational search algorithms and analysis tools for characterizing molecular conformations and flexibility |
Mesoscale & Statistical Tools
| Module | Description |
|---|---|
| Mesocite | Coarse-grained simulation for studying structural and dynamic properties of nano-to-micron-scale materials |
| MesoDyn | Classical density functional method for studying long-range and long-time-scale behavior of complex polymer systems |
| PhaseField | Predicts microstructure of hard materials — solidification and grain growth simulations |
| QSAR / QSAR Plus | Quantitative structure-activity relationships with extensive descriptors and GFA genetic algorithm; adds DMol3 descriptors and neural networks |
| Synthia | QSPR calculations for homopolymer and copolymer properties, rapid screening of candidate polymers |
Analysis & Crystallography Tools
| Module | Description |
|---|---|
| Morphology | Predicts crystal morphology from atomic structure — crystal shape, surface stability, additive and solvent effects |
| Polymorph Predictor | Searches for low-energy lattice energy minima across all reasonable space groups to predict potential polymorphs |
| Reflex / Reflex QPA | Simulates X-ray, neutron, and electron powder diffraction patterns; determines crystal structures from powder diffraction data with quantitative phase analysis |
| X-Cell | Efficient indexing algorithm with extinction-specific dichotomy procedures for powder diffraction data |
| Motif | Analyzes connectivity (hydrogen bond topology) in molecular crystals, interfaces with CCDC Mercury |

Materials Studio multiscale materials simulation — from quantum mechanics to mesoscale molecular visualization and property prediction
Key Force Fields
| Force Field | Type | Coverage |
|---|---|---|
| COMPASS III | Class II ab initio | Broadest coverage — organic/inorganic molecules, polymers, metals, metal oxides, gases |
| CVFF | Consistent valence force field | Peptides, proteins, amino acids, hydrocarbons |
| PCFF | Polymer consistent force field | Optimized for polymers and organic materials |
| Dreiding | Generic | Organic, biological, and main-group inorganic molecules |
| Universal | Generic | Rule-based force field covering the entire periodic table |
| MACE | Machine learning force field | New (2025) — quantum-level accuracy at classical MD cost; GPU accelerated; fine-tuning protocol added in 2026 |
| ReaxFF | Reactive force field | Bond-order-based reactive force field supporting continuous bond formation/breaking |
Technical Specifications
| Client Platform | Microsoft Windows 10/11 (64-bit) |
| Server Platform | Red Hat Enterprise Linux (RHEL) |
| Cloud Option | Dassault Systèmes 3DEXPERIENCE platform SaaS |
| GPU Acceleration | NVIDIA GPU (RTX A6000, RTX 4080 certified); Forcite Plus, CASTEP, and other modules |
| HPC Support | PBS Professional and SLURM job schedulers |
| Scripting / API | MaterialsScript API (Perl + Python, Python added in 2026) |
| Workflow Automation | BIOVIA Pipeline Pilot integration (Materials Studio Collection) |
Application Areas
Pharmaceuticals
Crystal structure prediction, polymorph screening, formulation optimization, drug stability, co-crystal design
Chemistry & Catalysis
Catalyst design, reaction mechanisms, surface chemistry, selectivity optimization, poisoning analysis
Polymers & Composites
Polymer blends, mechanical properties, aging, glass transition temperature, permeability, additive compatibility
Energy & Batteries
Li-ion battery materials, solid-state electrolytes, fuel cell membranes, hydrogen storage materials, solar cell materials
Electronics & Semiconductors
Band structure, electronic properties, defect calculations, doping studies, dielectric materials
Metals & Alloys
Phase diagrams, grain structure prediction, mechanical properties, corrosion, solidification
Nanomaterials
Carbon nanotubes, graphene, metal-organic frameworks (MOFs), zeolites, nanoparticle properties
Aerospace & Automotive
High-performance alloys, composite materials, thermal barrier coatings
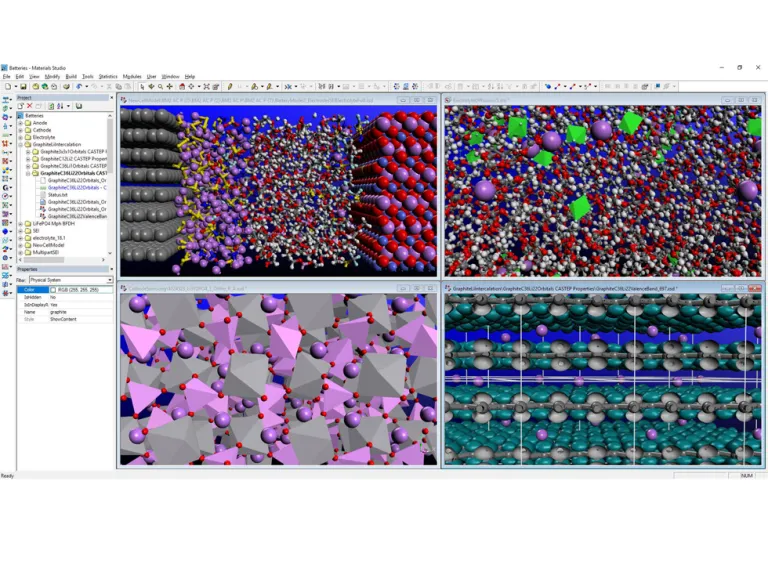
Materials Studio battery material simulation — multi-window molecular structure visualization and property analysis workspace

Materials Studio quantum mechanics toolkit — including CASTEP, DMol3, VAMP, and other density functional theory solvers
Key Competitive Advantages
Broadest Multiscale Coverage
The only platform spanning quantum, atomistic, mesoscale to microstructure in a single integrated environment
Comprehensive Crystallography Tools
Polymorph Predictor, Morphology, Reflex, X-Cell — unmatched in pharmaceutical and materials crystallography
COMPASS III Force Field
The most extensively validated Class II force field with the broadest material coverage
MACE Machine Learning Potential
New in 2025 — quantum-level accuracy at classical MD speed; fine-tuning protocol added in 2026
Enterprise Integration
Pipeline Pilot workflow automation + 3DEXPERIENCE platform supports global team collaboration
Recent Updates
| Date | Update |
|---|---|
| November 2025 | Materials Studio 2026 — MaterialsScript adds Python scripting support, MACE fine-tuning protocol, Adaptive Compressed Exchange (ACE) accelerating CASTEP hybrid functionals, GULP 6.4, ONETEP 7.3.95, Forcite GPU optimization |
| November 2024 | Materials Studio 2025 — MACE machine learning potentials (MACE-OFF23 organic molecules, MACE-MP-0b inorganic materials), MS Martini 3 coarse-grained force field, FlexTS transition state search (growing string method) |
| Late 2023 | Materials Studio 2024 — DMol3 Effective Screening Medium (ESM), DFTB+ GPU acceleration, Forcite dielectric analysis, ReaxFF SEI2021 |
Related Products
Learn About Materials Studio
Discover how multiscale materials simulation can accelerate your materials R&D and innovation
Contact Us